

Other Categories
UPDATE: OpenBiome has been permitted to continue distributing FMT to patients with severe, fulminant C. diff until December 31, at which time a new judgment will be made regarding the treatment’s availability.
You may have heard in recent weeks that OpenBiome, the largest distributor of fecal microbiota transplant (FMT) products in the United States, announced that it would suspend distribution following the end of enforcement discretion by the Food and Drug Administration (FDA).
Originally, their operations were expected to discontinue sometime in mid-September, but they have been permitted to distribute FMT through the end of this month (October 31, 2024). While this situation is unfolding, and we continue to work to resolve it to protect FMT access for our vulnerable patients, we are sharing this update to clarify the situation for all the C. diff survivors, patients, providers, and caregivers we represent.
What is the FDA’s current stance on FMT?
The previously mentioned ‘enforcement discretion’ is a special rule that allows access to a treatment method without needing to meet the stricter requirements that standard FDA-approved therapies must usually satisfy. This is typically because the therapy is low-risk and stands to positively impact public health. The FDA reserves the right to stop the use of these therapies.
Ending enforcement discretion for FMT means that healthcare providers looking to use FMT must now do so under the rules included in the finalized guidance on FMT released by FDA in 2022. An update to the draft guidance FMT had been under since 2013, and following the approval of fecal microbiota live-jslm (brand name RebyotaTM), this final document required that any continued use of FMT must be done under an ‘investigational new drug,’ or IND, application. INDs are what pharmaceutical developers use to act in accordance with federal laws on the distribution or use of new drugs across state lines before they gain market approval.
Why did OpenBiome decide to discontinue its FMT distribution?
After the October 31st deadline mentioned above, FDA will need to issue a decision based on the submission of OpenBiome’s IND application or other written permission to continue distributing FMT. As OpenBiome has stated publicly, the discontinuation of FMT distribution is not related to the safety or quality of the products but rather the difficulties the organization has had in making its IND submission over the past 22 months.
It is also worth noting that OpenBiome receives the FMT products it distributes nationwide from its partner, the University of Minnesota. UofM currently operates under its own IND for FMT use, which has been sanctioned by the FDA.
If the University of Minnesota has an IND, why does OpenBiome need one, too?
There has been much back-and-forth regarding this issue between UofM, OpenBiome, and FDA. In broad strokes, OpenBiome has been asked by FDA to submit a commercial IND–different from the research category of IND UofM operates under.
If we have FDA-approved microbiome therapies, why do we still need FMT?
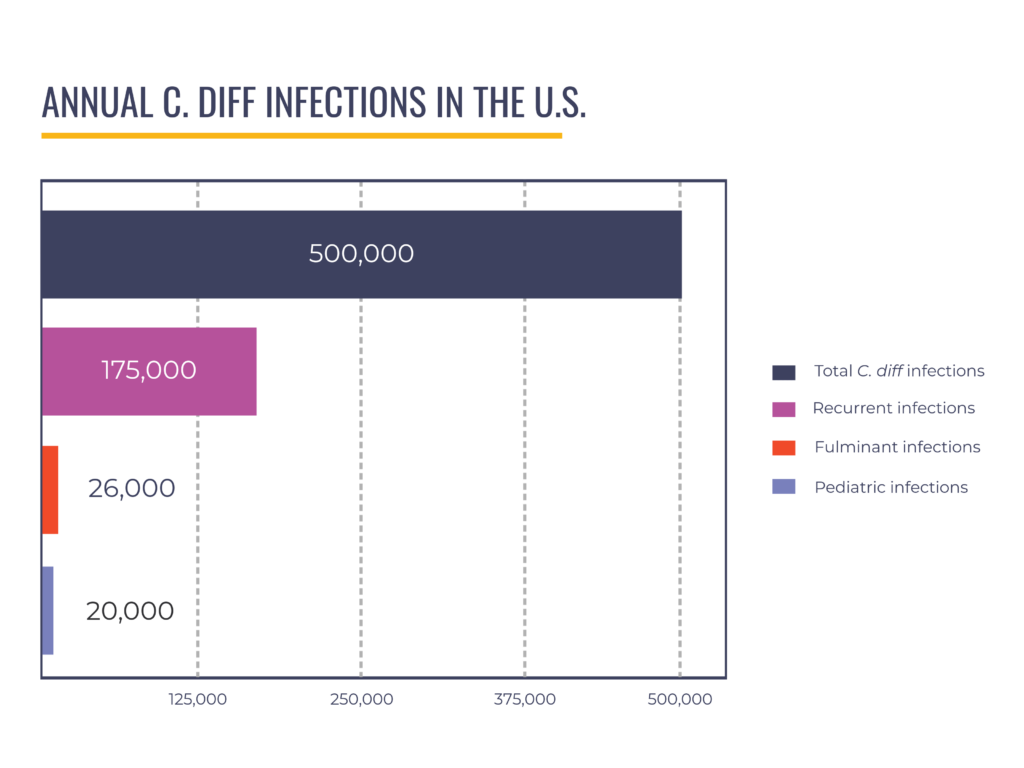
We have partnered with Industry to recruit for clinical trials and advocated for approval of fecal microbiota live-jslm (brand name RebyotaTM), the first live biotherapeutic product for recurrent CDI, which preceded approval of fecal microbiota spores live-brpk (brand name VowstTM). Some of our advocates have served as advisors to Industry and appeared in promotional materials to raise awareness of the new C. diff treatments. We believe the FDA’s approval of LBPs for recurrent CDI is a huge win for patients. However, these products have limitations that create a gap in care for specific patient populations; they are not indicated–meaning the products’ use is not currently recommended by FDA–for pediatric patients (around 20,000 annual cases) and those with severe, fulminant disease (up to 8% of hospitalized C. diff patients).
Recently updated clinical practice guidelines from the American Gastroenterological Association recommend conventional FMT over the new live biotherapeutics for mildly-to-moderately immunosuppressed patients and those with liver disease. This isn’t to say the therapies are unsafe for these patients, but there simply isn’t yet enough evidence for us to be sure of how safe or effective they may be in persons with those background conditions. Further, some patients cannot tolerate an LBP delivered via enema, and some, such as those with kidney disease, may be unable to undergo the bowel prep required for other LBPs.
If OpenBiome closes, where can C. diff patients in need get FMTs?
If OpenBiome were to cease operations completely, patients’ ability to access FMT would be severely hampered. However, other stool banks may still be accessible in certain locations. While FDA guidance at this point still allows individual healthcare institutions to process and administer FMTs, this is a heavy administrative and financial burden that limits FMT access to patients in and around major medical centers or teaching hospitals.
The other option is donor-directed stool transplants. While more accessible to patients outside the vicinity of facilities with stool banks, this method requires expensive testing of a single individual, which takes time and is primarily not covered by insurance. A C. diff patient who is going into septic shock does not have two or three days for testing to come back, assuming the selected donor is cleared.
What is PLF doing about the situation?
PLF has meticulously considered our position in the past few weeks, with the help of our Board members and advisors, most notably recent Board addition and retired regulatory attorney Debbie Trinker. We firmly believe that it is in the best interests of the most vulnerable C. diff patients to see FMT continue to be made available through OpenBiome. While PLF is treatment-agnostic, the efficacy of FMT and its availability for patients for whom FDA-approved therapies are untested or not indicated make it an essential option.
Since early in our founding, PLF has endeavored to help the FDA understand the experience of recurrent C. diff patients through participation in the Agency’s 2016 and 2019 public hearings on FMT, the September mentioned above 2022 Advisory Committee hearing, and – more recently – through a Patient Listening Session in March 2023. We have also regularly lobbied Congress to ensure the Agency is well-resourced.
The future of FMT is uncertain. However, we believe this treatment gap is unintentional, so PLF has been communicating with the FDA to preserve access to FMTs as a treatment option for C. diff patients. We requested a meeting with the FDA’s Center for Biologics Evaluation and Research (CBER) and the Agency was kind enough to grant us a meeting quickly, held on Friday, October 11, 2024. PLF staff, advocates, and advisors met with FDA officials involved in the matter. Patients shared stories regarding access (or lack thereof) to safe and effective FMTs, and clinicians shared data about the needs of the patients they treat. The meeting was well-attended, and we are optimistic about the FDA’s consideration of vulnerable C. diff patients moving forward.
As FDA-approved LBPs become more widely used, we hope people with recurrent C. diff will be offered them earlier. We also hope that more narrow-spectrum antibiotics are used for C. diff and other infections. Products are also in development that may prevent C. diff in vulnerable populations. Still, there remain patients who rely on FMT as an often life-saving treatment of last resort. We are hopeful that FDA will grant continued distribution of FMT products for pediatric and severe, fulminant patients until new or existing therapies are indicated for these populations.
If you or a loved one is a C. diff survivor who relied on FMT, you can respectfully write to the FDA to share your perspective at ocod@fda.hhs.gov and cberombudsman@fda.hhs.gov.
Attendees of the October 11 Meeting:
FDA Participants
Lorrie McNeill Director, Office of Communication, Outreach and Development
Susan Frantz-Bohn Deputy Director, Office of Communication, Outreach and Development
Peter Marks Director, Center for Biologics Evaluation and Research
Julie Tierney Deputy Director, Center for Biologics Evaluation and Research
David C. Kaslow Director, Office of Vaccines Research and Review, Center for Biologics Evaluation and Research
Karin Bok Deputy Director, Office of Vaccines Research and Review
Maureen Hess Health Science Advisor, Office of Vaccines Research and Review
Jay Slater Director, Division of Bacterial, Parasitic and Allergenic Products
Michelle Adams Senior Intergovernmental Affairs Specialist, Intergovernmental Affairs, Office of the Commissioner/Office of Policy, Legislation, and International Affairs
Theresa Finn Associate Director for Regulatory Policy
PLF Participants
Christian John Lillis Chief Executive Officer, Peggy Lillis Foundation
Debbie Trinker* Retired, Senior Vice President of Regulatory Affairs, Kemin Health LC
Bruce Hirsch* MD, Infectious Diseases Physician, Northwell Health
Stacy Kahn** MD, Director, Fecal Microbiota Transplantation, Boston Children’s Hospital
Carol Raye* Advocate and C. diff Survivor
Pam McCollister Advocate and C. diff Survivor
Maryann Webb* Advocate and C. diff Survivor
Kee Kee Buckley Advocate and C. diff Survivor
Erin Burns Executive Assistant, Peggy Lillis Foundation
Jamie Webb Program Manager, Peggy Lillis Foundation
Dan Freedberg** MD, Gastroenterologist, Columbia University Irving Medical Center
Vincent Young* MD, PhD, Professor of Microbiology and Immunology, University of Michigan
*PLF Board Member
**PLF Scientific Advisor



Hi there,
I want to thank you for this information.
I am a recent recipient of a FMT and survivor of C-diff.
My first experienced with C-diff was 2018 after a 3 cm polyp was removed from my ascending colon. Antibiotics were given before the procedure. Treatment consisted of two rounds of Vancomycin.
My second experience was Feb 4 2024. Treatment with Vancomycin 14 days treatment was started, 5 days after stopping Vancomycin diarrhea started, hospitalized 1 week, sent home with tapering Vancomycin treatment which continued until June 2024 after multiple trips to ER for extreme diarrhea and dehydration. Again hospitalized, FMT was performed, no improvement, a second FMT was performed, little improvement, one week later a third FMT performed. Finally, the diarrhea stopped. I was kept in the hospital from the first FMT to the third; three weeks. All transplants were performed via colonoscopy. I am happy to report I am doing so much better however, the thought of what can happen if I ever need another antibiotic is terrifying. Both times I had C-diff was caused by antibiotics. I’m told I am a carrier of C-diff and avoid PPIs which I had taking for over 14 years for acid reflux.
I hope the option for FMT via colonoscopy will always be available because I may need to have another one at some point in my life. I know there are other treatments but I am not a candidate. I have difficulty swallowing pills. I was hopeful FMTs would be available in more cities and areas. I didn’t have the help from my doctors in guidance on how or who’s responsible for finding a doctor or facility that performs them. I was totally one my own trying to find where I could go to have a FMT. Finally when I did, I had to wait 3 months to receive the procedure after being admitted into the hospital for severe diarrhea and dehydration. Again, it took 3 FMTs to cure me of C-diff although I am a carrier.
Lastly, I found caregivers in the hospital setting and medical offices settings don’t know what C-diff is or how take care of an infected patient. I also found through my experiences there are way for a patient to help with containing the spread of infection.
Again, I appreciate this information and your newsletter. I pray more awareness for C-diff. It’s so important and can save lives!
Thank you,
Mary
Thank you so very much for sharing your story. I have learned so much. I have had c.diff three separate times since November. Been on vancomycin each time with no relief. Medication is so expensive too as you know. Starting to give up hope of ever feeling better again.
Any chance of FMT ever being approved for other illnesses, in the USA?